При создании волокнистых катализаторов с высокой активностью
возникает ряд проблем, от решения которых зависит качество катализатора и
области его применения. Прежде всего это каталитическая активность металла и
прочность его закрепления на волокне, механическая прочность самого волокна и
стабильность сформированного катализатора.
Все указанные проблемы связаны с подбором условий модификации
исходного полимера, используемого в качестве волокнистого носителя, и технологии
закрепления на нем металла. Практически безальтернативным исходным материалом
для волокнистых катализаторов является сополимерное полиакрилонитрильное (ПАН)
волокно [1-5]. Именно этот полимер, благодаря наличию реакционноспособных
нитрильных групп, позволяет проводить направленную структурную модификацию в
ходе полимераналогичных превращений [3] и, таким образом, дает возможность
получения полидентатного лиганда, с которым в ходе дальнейшей трансформации
может координироваться атом металла.
Достаточно универсальны железосодержащие волокнистые
катализаторы [1, 5, 6]. Принципиальные технологические вопросы формирования на
ПАН волокне активных центров из ионов трехвалентного железа решены. Однако
требуются более детальные исследования процессов формирования на ПАН волокне
функциональных групп-аддендов, которые реализуются обработкой исходного волокна
водным раствором смеси солянокислого гидроксиламина и солянокислого гидразина с
последующей обработкой в водных растворах NaOH и FeCl3-6H3O.
Целью наших исследований явился поиск параметров создания
Fe3+-содержащего катализатора, обеспечивающего оптимальные, с точки зрения
практики, активность и стабильность катализатора при сохранении приемлемой
механической прочности ПАН волокна.
Исследования структурных трансформаций полиакрилонитрила в
процессе модификации проводились нами на промышленно выпускаемой ПАН комплексной
нити, полученной по роданидному способу из тройного сополимера акрилонитрила
(92.3%), метилакрилата (6.2%), итаконовой кислоты (1.5%), и моделировались на
ПАН пленках.
В ходе предварительных исследований варьировались соотношение концентраций
солянокислых гидразина и гидроксиламина (0.5-1), рН модифицирующего раствора
(2-10), продолжительность выдержки в нем (0.25-2 ч) и температура обработки
(100-150°С). Температура обработки в водном растворе NaOH составляла 95-100°С, продолжительность 30-120 с.
Заметим, что варьирование условий модификации приводит к тому, что указанные
группы - лиганды входят с разным статистическим весом в состав конструируемого
полимерного носителя. От их соотношения существенно зависит прочность
закрепления Fe3+ и стабильность катализатора [5]. По результатам этих
исследований были выбраны оптимальные параметры модификации ПАН волокна,
наиболее полно обеспечивающие указанные выше требования.
Приведем основные параметры постадийной модификации: соотношение концентраций
солянокислых гидразина и гидроксиламина 0.715, температура 110-130°С, рН = 6,
продолжительность обработки связана с температурой зависимостью
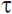 =
1.85-105 exp(-0.072t), обработка водным раствором NaOH в течение 30 с, выдерживание в
5%-ном водном растворе хлорного железа - 2 ч. =
1.85-105 exp(-0.072t), обработка водным раствором NaOH в течение 30 с, выдерживание в
5%-ном водном растворе хлорного железа - 2 ч.
Исследование полимерного носителя и готового катализатора, полученного по
указанным параметрам, проводилось методами электронной и оптической микроскопии
на растровом электронном микроскопе JSM-35 CF (JEOL), рентгеновском
микроанализаторе энергодисперсионного типа Link 860 (Link) с установкой
напыления JFC-1100 (JEOL) и EXAFS-спектроскопии (Extended x-ray Absorption fine
Structure - дальняя (протяженная) тонкая структура рентгеновских спектров
поглощения). Спектры ПАН пленок и волокон регистрировали на приборах IR
Spektrometer 5 DX (Nicolet) с программным обеспечением Hyper IR 1.57. Для
исследования элементного состава использовали фотоэлектронную спектроскопию на
приборе PHI 5400 (Perkin Elmer, USA). Исследования механической прочности
волокон до и после поэтапных обработок ПАН волокна проведены на универсальной
установке INSTRON-1122 со скоростью нагружения в пределах 0.05-1000 мм/мин в
диапазоне нагрузок от 10-3 до 5*103 Н с погрешностью не более 3-5% [7].
Напомним, что в ходе реакции ПАН с гидразином и гидроксиламином на волокне
образуются гидразиновые и гидроксамовоамидные группы, а обработка в кипящем
водном растворе NaOH приводит к появлению карбоксилат-ионных групп [8, 9].
Таким образом, в качестве функциональных групп-лигандов на волокне присутствуют
амино-, имино-, амидные и карбоксилат-ионные группы, и именно они являются
электронодонорными центрами, которые взаимодействуют с независимыми d-уровнями
атома железа.
Заключение о структуре носителя и механизмах протекания реакций модификации
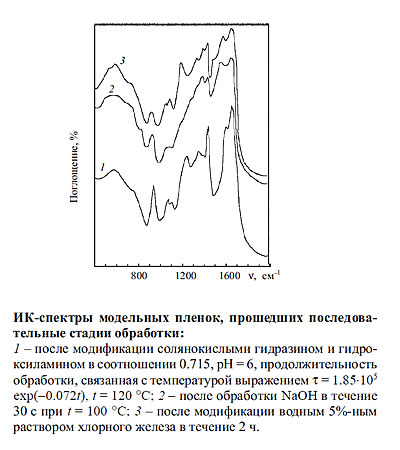
исходного ПАН были сделаны на основе изучения ИК-спектров модельных пленок,
регистрируемых после каждого этапа модификации.
Обработка ПАН солянокислыми гидразином и гидроксиламином в соотношении 0.715
при температуре 120°С и рН=6 раствора приводит к тому, что в ИК-спектрах
образцов (рисунок) появляются полосы, которые могут быть отнесены к поглощению
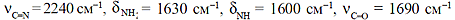 амидных
групп, входящих в состав следующих фрагментов: амидных
групп, входящих в состав следующих фрагментов:
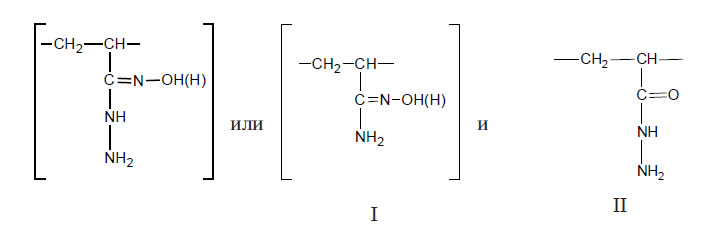
Заметим, что при раздельном либо одновременном повышении концентраций,
температур или продолжительности выдержки ПАН волокна в растворе не происходит
увеличения доли структур типа II. При оптимальном режиме обработки наблюдается
накопление структур типа I, часть которых может затем превращаться в
структуру II, а другая часть образует немногочисленные сшивки типа
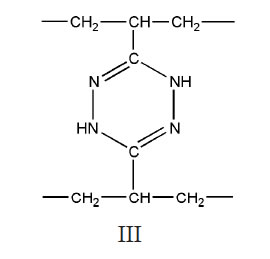
В ИК-спектрах отчетливо проявляются полосы поглощения гуанидиновой группы: 1662
и 1646 см-1, а также vNH гуанидиния в области 3300-3200 см-1.
На присутствие оксимных групп в составе структур типа I
указывает поглощение в области 3500 см-1, 1670 см-1(vOH) и отчетливо выраженная полоса 940 см-1,
соответствующая поглощению vNO [10]. Обратим внимание на то, что при
оптимальных режимах обработки в полимере сохраняется большое количество
неизменных нитрильных групп.
На следующей стадии обработки образца - кипячении в водном
растворе NaOH при 100°C в течение 30 с - продолжается образование структур типа II с одновременным
омылением их до карбоксилат-ионных групп
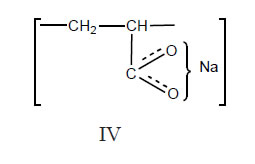
поглощение которых регистрируется в ИК-спектре образцов 1590 и 1390 см-1 (vs и
vas). Важным обстоятельством на этой стадии модификации является то, что не принявшие участия в превращениях на предыдущем этапе обработки группы CN
вступают в реакцию циклизации [3].
Нами установлено, что при извлечении образца из щелочного раствора на воздух
подобные циклические структуры присоединяют кислород, образуя N-оксиды.
Интенсивные полосы поглощения N-оксидов регистрируются в спектрах образцов в
области 1270-1260 см-1, что согласуется с данными [11]:
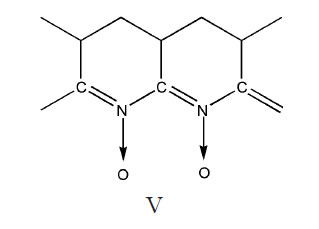
 В результате исследования структурных трансформаций установлено, что в ходе
описанных реакций образуется модифицированный полимер на основе ПАН, в составе
которого одновременно находятся структуры II, III, IV и V, содержащие центры
возможной координации с ионом переходного металла - атомы N и O и
функциональные группы, способные образовывать с металлом ковалентные связи. В результате исследования структурных трансформаций установлено, что в ходе
описанных реакций образуется модифицированный полимер на основе ПАН, в составе
которого одновременно находятся структуры II, III, IV и V, содержащие центры
возможной координации с ионом переходного металла - атомы N и O и
функциональные группы, способные образовывать с металлом ковалентные связи.
Как показывают результаты рентгеноструктурного анализа EXAFS и данные
электронной микроскопии, ионы Fe3+ располагаются по поверхности волокна и их ближайшими соседями являются два атома кислорода и три атома азота. При этом
на поверхности волокнистого носи¬теля регистрируются активные каталитические
центры, состоящие из 1, 2 и 3 атомов Fe. По оценкам, расстояние Fe-O и Fe-N
составляет 1.994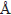 , а Fe-Fe - 2.98. , а Fe-Fe - 2.98.
Спектральные исследования в ИК-области показали, что у образцов, содержащих
железо, полоса N->O оксидов смещена в низкочастотную область на ~50 см-1 и
становится более широкой (см. рисунок). Э то согласуется с заключением 12] о
том, что по группам N->O идет координация металла. Поскольку в ближайшем
окружении Fe3+ определяется, как указывалось выше, два атома кислорода, то
можно предположить, что это атомы кислорода, входящие в состав циклических
N-оксидов. При координации с ними Fe3+ возможно образование довольно прочного
6-членного хелатного цикла.
Спектральные изменения в области поглощения амино- и иминогрупп в полилиганде
также свидетельствуют о координации с ними ионов железа. Атомы азота этих
функциональных групп занимают 3, 4 и 5 координационных положений иона Fe3+. В шестом координационном положении, скорее всего, находится молекула воды. В
пользу существования аквакомплекса свидетельствует наличие в ИК-спектрах
металлсодержащего катализатора полосы 750 см-1, которую относят к поглощению
координированной с ионом Fe3+ воды [12].
Довольно большое расстояние Fe-Fe (2.98), по-видимому, указывает на то, что
полиядерные комплексы образуются в данном случае с участием промежуточного
атома, например кислорода, как это отмечено в работе [13].
-
Установлено, что, варьируя параметры модификации, можно регулировать создание
структурированных ячеек для металлических частиц и с их помощью обеспечивать
оптимальное функционирование каталитических систем.
-
Изучены механизмы формирования и возможное строение разнолигандных
Fe3+-содержащих комплексов, локализованных на поверхности волокнистого носителя.
|


