Твердофазная дополиконденсация очищенного от непрореагировавшего капролактама
и циклических олигомеров (демономеризованного) водной экстракцией полиамида-6
(ПА-6), протекающая при 433-483 К в вакууме или токе инертного газа в режиме
необратимой реакции, становится [1] важной технологической стадией получения
полимера, используемого в производстве нитей технического назначения и различных
изделий литьем под давлением.
Однако в этой температурной области должен иметь место и действительно
реализуется [2, 3] процесс дополнительной кристаллизации (посткристаллизации)
полимера, приводящий к уменьшению объема и, возможно, изменению структурной
организации аморфных областей ПА-6, в которых, собственно, и осуществляется
реакция дополиконденсации.
Как отмечалось нами ранее [3-6], принципиальное отличие твердофазной
поликонденсации, протекающей в аморфных областях частично кристаллического
полимера, от реакции в расплаве заключается в отсутствии в первом случае у
концевых групп макромолекул трансляционной подвижности. Поэтому реакция может
идти по существу только за счет групп, образовавших контактные пары при
кристаллизации расплава.
С кинетической точки зрения это равносильно признанию того, что твердофазная
поликонденсация протекает как псевдомономолекулярная реакция дегидратации таких
контактных пар, которая может осложняться различной “подготовленностью”
последних к осуществлению химического акта.
Такие реакции называются, как известно [7], полихронными, и в случае
необратимой реакции первого порядка кинетические кривые линеализуются в
координатах уравнения [8]
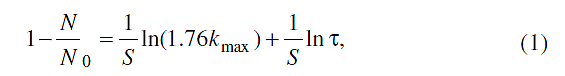
где N0 и N - начальное и текущее количества активных центров в реакционном
объеме; kmax - максимальная константа скорости; S - параметр кинетической
неэквивалентности этих центров, равный
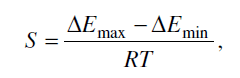
где ΔEmax и
ΔEmin - максимальное и минимальное значения энергии активации реакции.
Применительно к дополиконденсации полимера с начальной средней молекулярной
массой М0 и концентрацией монофункционального одноосновного [9] регулятора
молекулярной массы - [Рег]0 (моль на грамм демономеризованного полимера)
уравнение (1) трансформируется в уравнение *
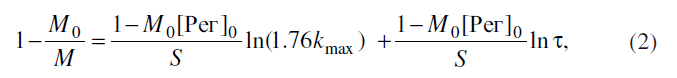
где М - текущее значение средней молекулярной массы полимера.
* Уравнение получено в предположении, что начальное число контактных пар равно
количеству концевых групп макромолекул, находящихся в недостатке [3].
Скорость мономолекулярной реакции не зависит, естественно, от объема
реакционного пространства, и поэтому само по себе уменьшение доли аморфных
областей ПА-6 в результате его посткристаллизации не может сказаться на скорости
дополиконденсации.
Однако нельзя отрицать возможности косвенного влияния на нее процесса
посткристаллизации через изменение фактического числа активных контактных пар и
степени их “подготовленности” к реакции, т.е. формально, через параметры [Рег]0
и S.
С другой стороны, если посткристаллизация сопровождается существенным изменением
объема (массы) аморфных областей ПА-6, то этим можно воспользоваться для
дополнительной проверки справедливости самого постулата о псевдомономолекулярном
характере химического акта при твердофазной дополиконденсации.
В качестве альтернативы этому постулату можно предположить, что специфика
твердофазной дополиконденсации определяется лишь тем, что обычная
бимолекулярная реакция протекает при переменном объеме (массе) реакционной
среды.
С целью выяснения, в какой мере сказанное относительно возможности влияния
посткристаллизации ПА-6 на кинетику его твердофазной дополиконденсации
справедливо, мы повторили описанные ранее [3] эксперименты по дополиконденсации
демономеризованного исчерпывающей экстракцией кипящим этанолом гранулята ПА-6
производства ОАО “Химволокно” (г. Щекино), получив одновременно кинетические
кривые изменения М и плотности полимера. При синтезе ПА-6 в качестве регулятора
молекулярной массы использовалась натриевая соль ɛ-аминокапроновой кислоты в
количестве 5.62-10-6 г/г полимера.
Реакция проводилась при 453-483 К и остаточном давлении ~ 67 Па. Среднечисленная
молекулярная масса полимера рассчитывалась из известного соотношения
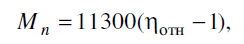
где ηотн - относительная вязкость раствора ПА-6 (1 г на 100 мл растворителя) в
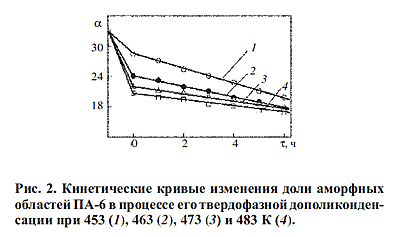
95.6%-ной H2SO4 при 298 К, а массовая доля аморфной фазы в полимере - из
соотношения
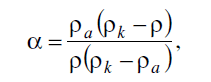
 где
ρk = 1230 кг/м3,
ρa = 1093 кг/м3 [10] - плотности кристаллических и аморфных
областей ПА-6 соответственно; ρ - фактическая плотность полимера, определенная с
точностью ±1 кг/м3 пикнометрическим методом в среде дистиллированной воды при
298 К. где
ρk = 1230 кг/м3,
ρa = 1093 кг/м3 [10] - плотности кристаллических и аморфных
областей ПА-6 соответственно; ρ - фактическая плотность полимера, определенная с
точностью ±1 кг/м3 пикнометрическим методом в среде дистиллированной воды при
298 К.
 Из кинетических кривых изменения
М и α, приведенных на рис.1 и 2, видно, что по
мере повышения температуры глубина и скорость протекания дополиконденсации
закономерно возрастают. Это касается как изотермических участков кинетических
кривых (τ > 0'), когда повышению температуры на 30 градусов соответствует
увеличение средней скорости реакции в ~1.5 раза, так и, особенно, на
неизотермических участках (0 < τ < 0' -
двадцатиминутный период подъема температуры гранул от комнатной до температуры
эксперимента), когда такому повышению температуры соответствует рост средней
скорости реакции в ~2.3 раза. Из кинетических кривых изменения
М и α, приведенных на рис.1 и 2, видно, что по
мере повышения температуры глубина и скорость протекания дополиконденсации
закономерно возрастают. Это касается как изотермических участков кинетических
кривых (τ > 0'), когда повышению температуры на 30 градусов соответствует
увеличение средней скорости реакции в ~1.5 раза, так и, особенно, на
неизотермических участках (0 < τ < 0' -
двадцатиминутный период подъема температуры гранул от комнатной до температуры
эксперимента), когда такому повышению температуры соответствует рост средней
скорости реакции в ~2.3 раза.
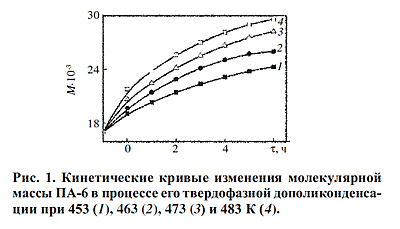
Аналогичная картина наблюдается и в изменении массовой доли аморфных областей
ПА-6. При 453 К на неизотермическом участке реализуется ~34% общего уменьшения
α, а при 483 К ~76%. На изотермическом участке процесса при всех исследованных
значениях температуры величина а уменьшается в соответствии с уравнением
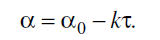
Воспользовавшись этим соотношением, получим выражение, позволяющее линеализовать
кинетическую кривую дополиконденсации в предположении, что изменению массы
аморфной фазы ПА-6 способствует обычная некатализируемая реакция прямого
полиамидирования при эквимольном соотношении амино- и карбоксильных групп и
исчерпывающем удалении из сферы реакции образующихся молекул воды.
Если масса аморфных областей Ga = αG , а количество молей в ней активных
концевых групп каждого вида изменяется в ходе реакции от N0 до N, то можно
записать:
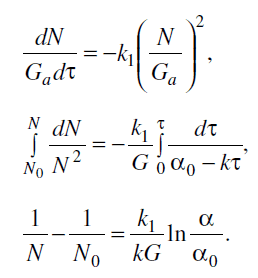
Приняв общую массу полимера G равной 1 г, получим

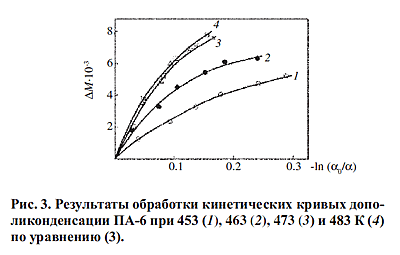 Результаты обработки кинетических кривых дополиконденсации по этому уравнению
(рис.3) показывают, что равенство не выполняется и, следовательно, предположение
о возможности реализации в условиях твердофазной дополиконденсации ПА-6 обычной
бимолекулярной реакции взаимодействия амино- и карбоксильных групп неверно. Результаты обработки кинетических кривых дополиконденсации по этому уравнению
(рис.3) показывают, что равенство не выполняется и, следовательно, предположение
о возможности реализации в условиях твердофазной дополиконденсации ПА-6 обычной
бимолекулярной реакции взаимодействия амино- и карбоксильных групп неверно.
В работе [5] подобный вывод был сделан на основе чисто формального анализа
кинетических кривых дополиконденсации.
Обратимся к уравнению (2) и проанализируем возможность использования его для
оценки влияния посткристаллизации на кинетические параметры дегидратации
контактных пар из амино- и карбоксильных групп при условии практической
невозможности прямого экспериментального определения концентрации таких пар,
потерявших способность к реакции в результате структурной перестройки аморфных
областей ПА-6.
Если исходить из того, что данный образец полимера характеризуется при комнатной
температуре некоторым распределением контактных пар по реакционноспособным
конфигурациям, то при мгновенном повышении его температуры до уровня, при
котором реакция становится возможной, реакционная система будет обладать
некоторым набором констант скоростей от kmax до kmin и соответствующим ему
параметром кинетической неэквивалентности S.
Очевидно, что при нормальном течении реакции набор констант скоростей должен
сужаться, а параметр S уменьшаться с увеличением
τ в результате исчерпания
наиболее активных контактных пар. Поэтому обработка экспериментальных данных по
уравнению (2) с последовательным перемещением точки отсчета начала реакции
τ0 в
сторону больших времен должна дать серию последовательно уменьшающихся величин
kmax и S. В случае же зависимости кинетических параметров реакции от каких-то
дополнительных факторов указанная последовательность должна, вероятно,
нарушаться.
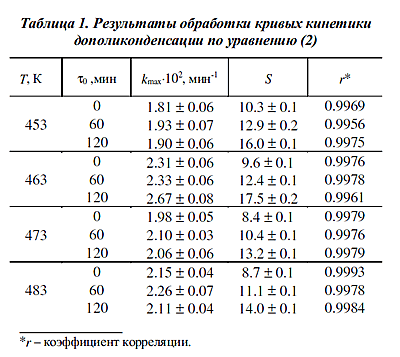 В табл. 1 приведены результаты расчета
kmax и S для всех исследованных значений
температуры при условии, что за М0 принимались значения, соответствующие моменту
0, 60 и 120 мин при неизменной величине [Рег]0 = 5.62-10-6 моль/г. В табл. 1 приведены результаты расчета
kmax и S для всех исследованных значений
температуры при условии, что за М0 принимались значения, соответствующие моменту
0, 60 и 120 мин при неизменной величине [Рег]0 = 5.62-10-6 моль/г.
Как можно видеть, адекватность уравнения (2) экспериментальным данным не зависит
от того, какая часть кинетической кривой дополиконденсации по нему
обрабатывается. Поэтому противоположная ожидаемой тенденция в изменении
параметров kmax и S может рассматриваться как аргумент
в пользу выдвинутого выше предположения о косвенном влиянии посткристаллизации на кинетику твердофазной дополиконденсации ПА-6.
Основываясь на полученных данных, следует полагать, что это влияние выражается в
дополнительном появлении как “удачных”, так и “не очень удачных” в кинетическом
плане конфигураций контактных пар. Однако нельзя, по-видимому, исключать и
возможности превращения части первоначально реакционноспособных контактных пар в
кинетически неактивные в результате фиксации увеличенных до критических значений
расстояний между образовавшими эти пары амино- и карбоксильными группами.
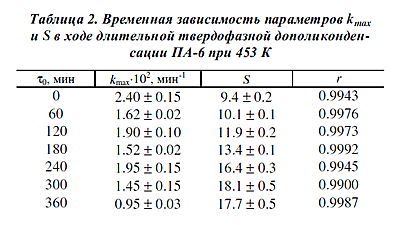 Достаточно четко выраженная тенденция к уменьшению прироста параметра S по мере
протекания реакции с повышением температуры вполне согласуется с замедлением при
этом процесса посткристаллизации (см. рис.1). Достаточно четко выраженная тенденция к уменьшению прироста параметра S по мере
протекания реакции с повышением температуры вполне согласуется с замедлением при
этом процесса посткристаллизации (см. рис.1).
Принципиальный интерес с рассматриваемой точки зрения представляют кривые
кинетики дополиконден- сации ПА-6, полученные при очень быстром нагревании
полимера до температуры реакции и относящиеся к длительным временным интервалам.
Такая кинетическая кривая (неизотермический участок реакции 5 мин, общая
продолжительность процесса 20 ч) приведена в работе [3], а результаты ее
обработки по обсуждаемой методике показаны в табл.2.
Из сопоставления цифр, приведенных в табл. 1 для 453 К и в табл.2, следует:
- процесс посткристаллизации не сказывается на адекватности уравнения (2) даже в
том случае, когда он развивается по сути параллельно реакции дополиконден- сации;
- в условиях длительного неизотермического режима успевают прореагировать
наиболее реакционноспособные контактные пары, с одной стороны, и появиться менее
реакционноспособные, с другой, что выражается в уменьшении
kmax и росте S при
обработке всей кривой кинетики дополиконденсации (τ0 = 0);
- при больших длительностях реакции, когда посткристаллизация, по-видимому,
завершается, изменение kmax и S в ходе реакции приобретает характер,
согласующийся с “нормальным” течением полихронной реакции.
Резюмируя изложенное, представляется правомерным утверждать, что развивающийся
параллельно твердофазной дополиконденсации ПА-6 процесс его посткристаллизации
влияет на ход этой полихронной реакции главным образом за счет увеличения
параметра, отражающего кинетическую неэквивалентность контактных пар,
образованных амино- и карбоксильными группами макромолекул. Устранение этого
эффекта было бы равносильно катализу твердофазной дополиконденсации ПА-6.
- На основе данных по кинетике изменения молекулярной массы и плотности
полиамида-6 при его нагревании в вакууме при 453-483 К обсуждается характер
влияния процесса дополнительной кристаллизации полимера на кинетические
параметры реакции его твердофазной дополиконденсации.
- Эта реакция является полихронной и сводится к псевдомономолекулярной
дегидратации контактных пар, образованных амино- и карбоксильными группами
макромолекул при кристаллизации расплава полимера.
|


